
Out-of-Specification (OOS) results are critical in the pharmaceutical industry as they directly relate to product quality, patient safety, and regulatory compliance. Regulatory agencies such as the USFDA (United States Food and Drug Administration) and MHRA (Medicines and Healthcare products Regulatory Agency) provide distinct guidelines to handle OOS results. While both aim to ensure compliance and product quality, their approaches vary in terms of investigation phases, documentation, and regulatory alignment.
This article highlights the differences between the USFDA OOS Guidance and the MHRA OOS Guidance, with a detailed comparison of their investigation phases and key focus areas.
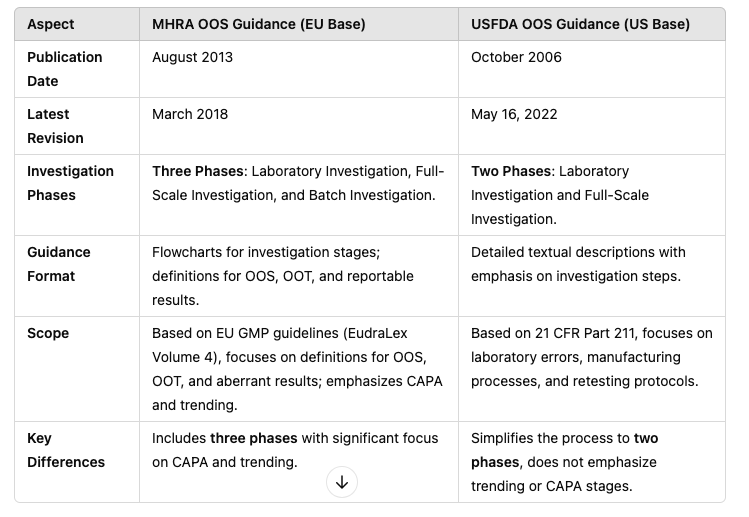
Overview of OOS Guidelines
—USFDA OOS Guidance
Title: “Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production”
Publication Date: October 2006, Last Revision : May 2022
Focus: Emphasizes a two-phase investigation process covering laboratory and manufacturing investigations.
—MHRA OOS Guidance
Title: “Guidance for the Interpretation of Out of Specification Results”
Publication Date: August 2013
Revision: March 2018
Focus: Outlines a three-phase investigation process with a strong emphasis on compliance with EU GMP standards and corrective actions.
Key Difference: Number of Investigation Phases
USFDA OOS Investigation Phases
The USFDA guidance defines a two-phase investigation process
Phase I: Laboratory Investigation
This phase involves investigating the possibility of laboratory errors, such as sample handling, instrument calibration, or analyst mistakes.
Key Actions:Review of laboratory test procedures, equipment logs, and calculations.
Cross-verification of results with raw data.
Limited retesting is allowed if errors are identified and justified.
Phase II: Full-Scale Investigation
If no laboratory errors are identified, the investigation extends to the manufacturing process.
Key Actions:Evaluate the entire production process, including raw materials, equipment, and environmental factors.
Perform root cause analysis for potential manufacturing deviations.
This phase determines the impact of the OOS result on other batches and future production runs.
MHRA OOS Investigation Phases
The MHRA guidance outlines a three-phase investigation process, offering more granularity:
Phase I: Initial Laboratory Investigation
Similar to the USFDA Phase I, this phase investigates laboratory errors.
Key Actions:Detailed review of testing methodology, equipment logs, and operator techniques.
Hypotheses for OOS occurrence are documented with clear conclusions.
Phase II: Full Investigation
Combines laboratory and manufacturing investigations.
Key Actions:Examination of the manufacturing process, raw materials, and sampling methods.
Identification of systemic issues or procedural deviations contributing to OOS results.
This phase is more comprehensive than the USFDA Phase II, emphasizing EU GMP alignment.
Phase III: Post-Investigation Analysis and CAPA
This phase is unique to MHRA and focuses on corrective and preventive actions (CAPA) based on investigation findings.
The phase 3 reviews the completed manufacturing investigation and combined laboratory investigation into the suspect analytical results, and/or method validation for possible causes into the results obtained. It’s a conclusive phase.The investigation report should contain a summary of the investigations performed; and a detailed conclusion.
Key Actions:Evaluate trends in OOS results to identify recurring issues.
Implement system-wide CAPA to prevent future OOS results.
Align investigation outcomes with quality management systems and EU GMP requirements.
Critical Focus Areas in Each Guidance
USFDA Key Focus Areas
- Systematic Laboratory Investigation: Detailed root cause analysis to identify laboratory errors.
- Manufacturing Process Examination: Investigate production-related issues if no lab errors are found.
- Batch Disposition: Decisions on batch acceptance or rejection are based on scientifically justified conclusions.
- Statistical Tools: Appropriate use of statistical methods for retesting and resampling.
MHRA Key Focus Areas
1. CAPA Implementation: Focus on preventing recurrence of OOS results.
2. Three-Phase Investigation: A structured approach covering laboratory, full-scale, and CAPA phases.
3. Documentation and Audit Trail: Heavy emphasis on recording every step in line with EU GMP requirements.
4. Trend Analysis: Identifying patterns in OOS results to improve processes.
Conclusion
The USFDA OOS guidance and MHRA OOS guidance share the same fundamental goal of ensuring product quality and regulatory compliance, but their approaches differ significantly. The USFDA guideline adopts a two-phase investigation process with a strong focus on identifying laboratory and manufacturing issues. In contrast, the MHRA guidance introduces a third phase, emphasizing systemic improvements through CAPA and trend analysis, aligning with EU GMP principles.
Pharmaceutical companies operating globally must adapt to these differences to maintain compliance with both agencies. A robust understanding of these guidelines is essential for effective quality assurance and regulatory adherence in a competitive and highly regulated industry.






кайт школа хургада
Read the latest world sports news: football, hockey, basketball, MMA, tennis and more. Insiders, forecasts, reports from the scene. Everything that is important for sports fans to know – in one place.
very useful information.
Thank you!!